Los resultados de los ensayos clínicos son el estándar que permite generar conocimiento y mejorar los patrones del cuidado médico. Suponen, por lo tanto, un motor de la actividad científico-técnica en el campo de la medicina. Un sistema de salud como el español, bien estructurado, es una ventaja para la realización de estos ensayos. El alto nivel científico-técnico de nuestros profesionales se traduce, además, en una llamada para que se realicen más ensayos clínicos en España.
Los ensayos clínicos constituyen una oportunidad de acceder precozmente a determinados tratamientos especialmente en pacientes que carecen de otras alternativas y para los que, en el caso de no poder acceder a los ensayos clínicos que se realizan en otros países, la espera hasta la autorización del medicamento podría suponer la pérdida de una última oportunidad.
Los ensayos clínicos se estructuran en torno a las siguientes fases:
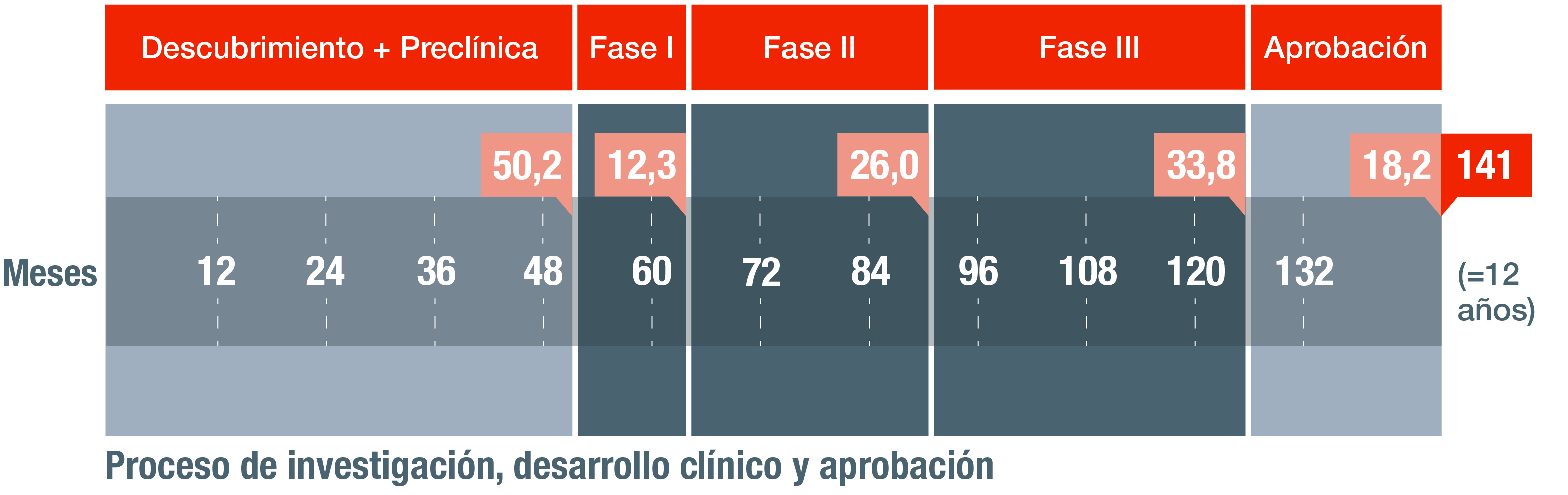
INV. Investigational New Drug. Paquete de información que se somete a los reguladores para la autorización de los ensayos clínicos en humanos. Debe contener los resultados preclínicos, estructura química, modo de actuación, toxicidad y efectos secundarios de los ensayos en animales y el proceso de producción del compuesto. Asimismo, debe describir la planificación de los ensayos clínicos en humanos: no de participantes, descripción detallada del ensayo, centros implicados, criterios de elección de pacientes, medidas de seguridad y eficacia, etc
Fase I. Incluye los primeros estudios que se realizan en seres humanos, pretenden demostrar la seguridad del compuesto y orientar hacia la pauta de administración más adecuada para estudios posteriores. Se trata de estudios de farmacología humana. Suelen realizarse en unidades de farmacología clínica utilizando alrededor de 20 a 100 sujetos por ensayo (voluntarios sanos y/o pacientes).
Fase II. Tiene como objetivo proporcionar información preliminar sobre la eficacia del producto y establecer la relación dosis-respuesta; son estudios terapéuticos exploratorios. Se realizan con un número limitado (100 a 300) de pacientes. Uno de los principales objetivos de este tipo de ensayos es determinar el rango de dosificación apropiado.
Fase III. Estos ensayos evalúan la eficacia y seguridad del tratamiento experimental en las condiciones de uso habituales y con respecto a las alternativas terapéuticas disponibles para la indicación estudiada. Se trata de estudios terapéuticos de confirmación. Se realizan en número elevado de pacientes (más de 1000) y suelen durar de 3 a 6 años.
Fase IV. NDA (New Drug Application) Solicitud para la comercialización de un nuevo medicamento. Se remite a las agencias reguladoras competentes. Contiene la información detallada recopilada a lo largo de todo el proceso de desarrollo. Debe presentar evidencia de que el nuevo fármaco tendrá el efecto deseado. Puede llegar a tener más de 120.000 páginas.
Postautorización. Ensayos clínicos Fase IV. Se realizan después de la comercialización del fármaco para estudiar condiciones de uso distintas de las autorizadas, como nuevas indicaciones. Estudios Post-autorización. Se realizan después de la comercialización del fármaco para estudiar la efectividad y seguridad en la utilización clínica diaria y otras cuestiones sobre el uso de fármacos en condiciones reales de práctica clínica (farmacovigilancia, farmacoeconomía, etc.).
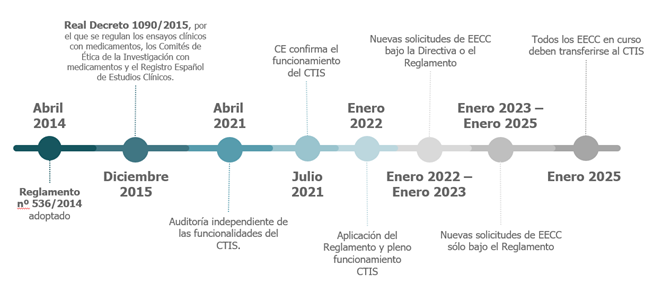
El 31 de julio de 2021, la Comisión Europea publicó en el Diario Oficial de la Unión Europea la Decisión 2021/1240, de 13 de julio de 2021, sobre la plena funcionalidad del portal del Sistema de Información de Ensayos Clínicos (CTIS, por sus siglas en inglés) y la base de datos de ensayos clínicos de la UE.
España es una potencia internacional en ensayos clínicos y ahora también está liderando el proceso de adaptación al nuevo Sistema de información de ensayos clínicos (CTIS). Nuestro país fue el primero de la Unión Europea en adoptar el Reglamento Europeo de Ensayos Clínicos – en el que se enmarca el CTIS – a través del Real Decreto 1090/2015, lo que ha supuesto que ya se haya ido implementando una simplificación y armonización de los procedimientos a nivel nacional.
La aplicación plena del Reglamento de Ensayos Clínicos (UE) nº 536/2014 en todos los países de la UE y del Espacio Económico Europeo (EEE) –Islandia, Liechtenstein y Noruega– tuvo lugar el 31 de enero de 2022 y, desde el 31 de enero de 2023, todas las solicitudes iniciales de ensayos clínicos deben presentarse de conformidad con el Reglamento EC y a través del Portal CTIS.
El objetivo del reglamento es crear un entorno favorable para la realización de ensayos clínicos en Europa y, al mismo tiempo, garantizar que se mantengan altos estándares de calidad de los mismos, que preserven la seguridad de los participantes.
La aplicación del Reglamento EC conllevará:
El CTIS apoyará los procesos de trabajo de los Estados miembros de la UE, los países del EEE y los promotores, durante todo el ciclo de vida de un ensayo clínico. Asimismo, proporcionará supervisión reglamentaria de los ensayos y herramientas para su seguimiento.
La Agencia Española de Medicamentos y Productos Sanitarios (AEMPS) proporciona, en un formato de preguntas y respuestas, información sobre los aspectos prácticos que conlleva la aplicación del Reglamento (UE) Nº 536/2014 y del Real Decreto 1090/2015ª través elaborado una serie de documentos de instrucciones, documentos vivos y en continua revisión para dar adecuada respuesta a las novedades. El documento es complementario del «memorando de colaboración» entre la AEMPS y los CEIm también público.
A continuación, puedes consultar en el siguiente enlace las diez razones por las que España es líder mundial en Ensayos Clínicos. Se puede encontrar también su traducción en inglés 10 reasons why Spain is a world leader in Clinical Trials.
Best es un proyecto estratégico impulsado, desde 2006, por la industria farmacéutica y en el que se integran todos los stakholders públicos y privados para crear una plataforma de excelencia en investigación clínica de medicamentos en España.